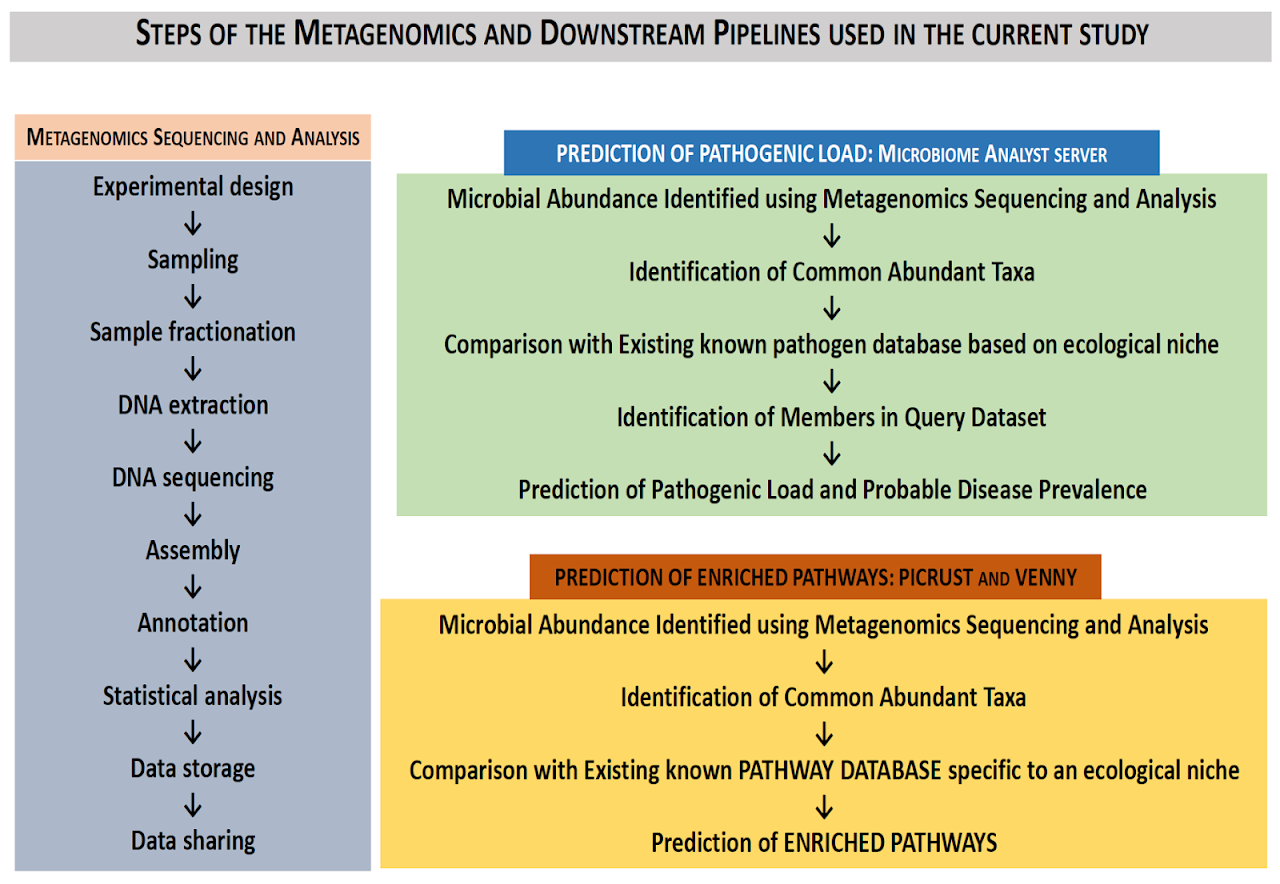
Analyses of Rhizospheric Metagenome Using Next Generation Sequencing
DNA extraction and QC
DNA was extracted from soil samples using our internal protocol (C. Yeates et. Al.). The quality of DNA was checked on 0.8% agarose gel (Figure 1) and quantified using QUBIT dsDNA HS kit. (Table 1: QUBIT quantification for the extracted sample) Purified sample was taken further for processing.
Design of PCR primers with multiplexing index and Illumina sequence adapters
PCR primers for amplification of V3&V4 regions of 16S rRNA gene were designed with appropriate sample barcoding index sequences and Illumina adapter sequences. Care was taken while designing these primers to allow multiplexing of samples but without allowing primer dimer formation or secondary structures that would reduce PCR efficiency. The primer sequences were used as given in Illumina protocol.
PCR amplification of target region
PCR conditions were optimised for each primer set that is different only by the barcoding indices. V3&V4 regions were amplified from the DNA samples and purified using Agencourt XP beads. The library was then quantified using UBIT dS DNA HS kit and normalized to 10 nMol/l for sequencing. The normalized library QC was done using an Agilent Bioanalyser DNA HS Chip. (Figure 2).
Sequencing
The genome sequencing reveals the order of DNA nucleotides (the order of As, Cs, Gs and Ts) in genome/ organisms. The quality and quantity of the prepared libraries met the Illumina standards required for further sequencing and hence the library was further sequenced on Illumina MiSeq plaform using 2X250 bp chemistry with generation of more than 1.5 million reads.
Bioinformatics Analysis
The quality of raw reads of Illumina sequencing was checked for the ambiguous bases, Phred score >Q30, read length, nucleotide base content and other parameters by using FASTQC toolkit (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The quality processed paired end reads were analysed with three different pipelines: QIIME, MEGAN and Kraken (along with in-house scripts), for a comprehensive and comparative analysis of the Rhizosphere soil metagenome data. QIIME data was further used to determine the gene composition and alpha rarefaction within the sample.
Read Summary and QC
The Quality control parameters suggest good quality reads. The sequence length distribution is at 250bp, there were no ambiguous bases present in the reads and the average Phred score was >30 for all the reads.
Table 2: Total PE reads of Sieved Sample
16s rRNA Metagenome Analysis
QIIME Analysis
The processed reads were clustered into OTU’s (Operational Taxonomic Units) by using QIIME software to identify the microbial community. These OTU’s were further used for taxonomic assignment, Phylogenetic, Diversity analysis and abundance estimation. QIIME (an abbreviation for Quantitative Insights Into Microbial Ecology) is a bioinformatics pipeline designed for analyzing microbial communities. The software clusters the marker gene nucleotide sequences into OTUs (Operational Taxonomic Units) and taxonomically annotates the OTUs by looking for sequences similar to them on a reference taxonomic database. Rarefaction curves are necessary for estimating species richness.
MEGAN Analysis
MEGAN analysis was started with comparing the reads with NCBI nr database, using RAPSearch(E value <= 0.001, Percent identity >=30). Then, MEGAN assigned a taxon ID to processed read results based on NCBI taxonomy which created a MEGAN file that contains required information for statistical and graphical analysis. Lastly, lowest common ancestor (LCA) algorithm was run to inspect assignments, to analyze data and to create summaries of data based on different NCBI taxonomy levels. LCA algorithm simply finds the lowest common ancestor of different species.
Kraken Analysis
Kraken program was additionally used for assigning taxonomic labels to metagenomic DNA sequences. Kraken’s classification algorithm performs mapping of k-mers to taxa by querying a pre-computed database. Kraken creates this database through a multi-step process, beginning with the selection of a library of genomic sequences. Kraken includes a default library, based on completed microbial genomes in the National Center for Biotechnology Information’s (NCBI) RefSeq database. The classification results can be visualized using Krona software.
Comparative Taxonomy Conclusion
The majority of the sequences analyzed by all three methods, belonged to the phylum Proteobacteria, which includes a wide variety of pathogens, such as Escherichia, Salmonella, Vibrio, Helicobacter, Yersinia, and many other notable genera. Others are free-living (nonparasitic), and include many of the bacteria responsible for nitrogen fixation. Most abundant class of Proteobacteria in the sample turns out to be Alphaproteobacteria by analysis of both QIIME and KRAKEN, which include agriculturally important bacteria capable of inducing nitrogen fixation in symbiosis with plants. The rhizosphere soil depicted the high abundance of nitrogen-fixing bacteria. Heliobacterium modesticaldum Ice1, was the most abundant of all the microbial community according to MEGAN analysis which is a well-known nitrogen-fixing bacterium.
Gene Content Prediction
PICRUSt workflow is used to predict the gene content of our metagenome. PICRUSt is designed to estimate the gene families contributed to a metagenome by bacteria or archaea identified using 16S rRNA sequencing. These gene content predictions are pre-calculated for protein-coding genes present in KEGG and 16S rRNA gene copy number. The QIIME OTU table was used to predict functions and the pathways using PICRUSt. Additionally, it was identified which OTUs were contributing to which function. First step was to correct the OTU table based on predicted 16S rRNA copy number for each organism in the OTU table. The functional predictions of KEGG Ortholog (KOs) were carried out using the corrected OTU table as input. Finally, the KO’s were collapsed to KEGG pathways (Table 6) because one KO can map to many KEGG Pathways. Additionally, PICRUSt was used to connect the OTUs that are contributing to each KO.
For the convenience of work the different plant samples were named BEC 1- BEC 7.
- BEC 1 – Acrostichum aureum Linn.
- BEC 2 – Drynaria quercifolia (L.) J. Sm.
- BEC 3 – Pyrrosia lanceolata (L.) Farwell
- BEC 4 – Marsilea minuta Linn.
- BEC 5 – Certopteris thallictroides (L.) Brongn.
- BEC 6 – Microsorum punctatum (L.) Copel.
- BEC 7 – Christella dentata (Forssk.) Brownsey & Jermy
The OTUs represent the operational taxonomic units. In each of the sequenced samples, OTU 1 represents the endophytic metagenome of Acrostichum, OTU2 represents the endophytic metagenome of Drynaria, OTU 3 represents the endophytic metagenome of Pyrrosia. OTU 4 represents the endophytic metagenome of Marsilea, It is collected from a partly submerged area. The OTU 5 represents the endophytic metagenome of Ceratopteris, This is a completely terrestrial fern found usually on the edges of water bodies. OTU 6 represents the endophytic metagenome of Microsorum, collected from the trunk of a tree and OTU 7 represents the endophytic metagenome of Christella, a terrestrial fern.
The first and second year study involved regular field trips for understanding and estimating the occurrence of the different fern species in the ‘non-core’ area of the Indian Sunderbans. Four different study sites were selected based on the presence of human settlements and abundance of fern species in the area.